|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Rubrik: Tagesberichte |
Print-Version
|
|
TGF-Beta als wichtiges Signal bei angeborener Krankheit DiGeorge-Syndrom weiter aufgeklärt | |
|
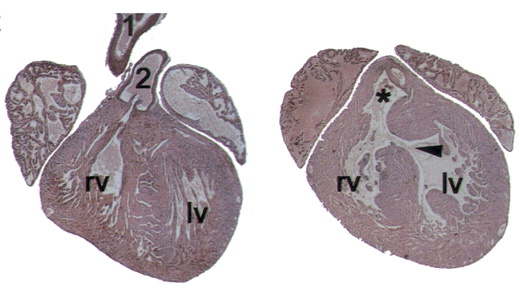
Wird die Signalkaskade mit dem Eiweiss TGF-Beta unterbrochen, dann führt das in Mäusen zu einem Krankheitsbild, das dem DiGeorge-Syndrom beim Menschen gleicht. Diese Erkenntnisse, die Forschende unter Leitung des ETH-Professors Lukas Sommer gewannen und im Wissenschaftsmagazin „Genes & Development“ publizierten, könnten die Grundlage für neue Therapien sein. Von Christoph Meier Fehlbildungen im Gesicht, Herzprobleme, Immunschwäche bedingt durch mangelnde Entwicklung des Thymus’ oder Unterfunktion der Nebenschildrüsen: Das sind einige der Charakteristika, die das DiGeorge-Syndrom bestimmen.Von dieser angeborenen Krankheit beim Menschen, die bei einer von 4000 Geburten lebender Säuglinge auftritt, weiss man auch, dass sie gewöhnlich verbunden ist mit einem Verlust eines kleinen Teils des Chromosoms 22. Das DiGeorge-Syndrom ist somit die häufigste Krankheit beim Menschen, die durch eine chromosomale Mikrodeletion ausgelöst wird. Aus der Embryonalentwicklung weiss man zudem, dass die beim DiGeorge-Syndrom betroffenen Gewebe von so genannten Neuralleisten-Zellen abstammen. Doch trotz des Wissens über die Chromosomen und die Zellen sind die molekularen Grundlagen der Krankheit weitgehend ungeklärt. Eine neue Forschungsarbeit unter der Leitung des ETH-Zellbiologen Lukas Sommer zeigt nun (1), dass die Signalkaskade mit dem Eiweiss TGF-Beta eine entscheidende Rolle spielt. Als die Forscher in den Neuralleistenzellen von Mäusen den Rezeptor von TGF-Beta inaktivierten, führte das bei den Tieren nach 18 Tagen der Embryonalentwicklung zu einem Krankheitsbild vergleichbar dem DiGeorge-Syndrom. So wiesen die Tiere unter anderem eine defekte Herzscheidewand (vgl. Bild), deformierte Schädelknochen oder einen reduzierten Thymus auf. Keinen Einfluss schien der unterbrochene Signalweg auf das Wanderverhalten der Neuralleistenzellen zu haben. So befanden sich diese Zellen beim gut zehn Tage alten Embryo am erwarteten Ort im Organismus. Danach, so legen es die Resultate nahe, entwickeln sie sich nicht mehr richtig weiter.
|
Da weder TGF-Beta noch sein Rezeptor durch den für das DiGeorge-Syndrom typischen Verlust auf dem Chromosom 22 betroffen sind, stellte sich Frage, welches andere Gen in dieser Region mit dem TGF-Signalweg verbunden sein könnte. Sommer und seine Kollegen fanden einen entsprechenden Kandidaten, Crkol. Dessen Proteinprodukt, CrkL, kann nur phosphoryliert und somit aktiviert werden, wenn eine TGF-Beta-Signalisierung erfolgt, wie die neuen Arbeiten demonstrierten. Das überraschte insofern nicht, als bekannt war, dass Mäuse mit einem mutiertem Crkol-Gen Charateristiken des DiGeorge-Syndroms aufweisen. Diese sind aber weniger ausgeprägt als beim Unterbruch der TGF-Beta-Signalkaskade. Insgesamt weisen die Resultate darauf hin, dass das Schicksal der Neuralleistenzellen zumindest in späteren Entwicklungsstadien unabhängig von CkL durch TGF-Beta beeinflusst wird. In einer Medienmitteilung des Wissenschaftsmagazins „Gene&Development“ (2), in dem die Forschung publiziert wurde, meinte Lukas Sommer zur Bedeutung der Forschung: „Unser Mausmodell liefert die molekulare Basis für die klinischen Befunde des DiGeorge-Syndroms. Es zeigt, dass die Regulierung des TGF-Signals eine entscheidende Rolle bei der Differenzierung der Neuralleistenzellen und den Ursachen der Krankheit spielt.“ |
||||||
|
Fussnoten:
Sie können zu diesem Artikel ein Feedback schreiben oder die bisherigen lesen. | |||||||